
 |
 |
 |
TS با سن مادر یا پدر مرتبط نیست. اگر چه موارد معدودی عود خانوادگی وجود داشته است ، TS معمولا تکگیر میباشد و خطر عود تجربی برای بارداریهای بعدی ، نسبت به جمعیت عمومی افزایش نمییابد. اگر چه بر پایه یافتههای سونوگرافی جنین مانند هیگروم کیستی شک به TS ایجاد شود، تشخیص باید تعیین کاریوتیپ پرزهای کوریونی یا آمینوسیتها تائید شود.
صرفا تعداد کمی بارداری در بین آن دسته از مبتلایان به TS که بطور خودبخود خونریزی قاعدگی داشتهاند، گزارش شده است. 3/1 فرزندانی حاصله ، دچار ناهنجاریهای مادرزادی مانند بیماری مادرزادی قلب ، سندرم داون و فقرات دو شاخه بودهاند. علت یا علل این خطر افزایش یافته برای ناهنجاریهای مادرزادی مشخص نشده است.
البته، همانطور که در ادامه میخوانید، وی با گردآوری و تفسیر دادههای به دست آمده از پژوهشهای دانشمندان دیگری، به این مفهوم پایهای دست پیدا کرد.
در پژوهشهای گرود، کمتر با طراحی آزمایش روبهرو میشویم. در حقیقت ، او با مشاهده و گردآوری اطلاعات توانست، فرضیههای خود را تا اندازهای اثبات کند. سرانجام، زیستشیمیدانها با پژوهش هایی که طی نیم سده انجام دادند، توانستند تصویر دقیقتری از اساس بیماری آلکاپتونوریا عرضه کنند.
در این مقاله، شما با شیوهی پژوهشی گرود آشنا میشوید و درمییابید که چگوه گردآوری دادهها و تفسیر آنها به یک یافتهی زیستشناسی میانجامد.
طرح مسأله
در سال 1898 میلادی، خانوادهای به دکتر گرود مراجعه کردند که ادرار کودکشان پس از برخورد با هوا ، تیره رنگ میشد ( پوشاک نوزاد در بخش جلو قهوهای تیره تا سیاه میشد). سه سال بعد، کودک دیگری در همان خانواده به دنیا آمد که به همین بیماری دچار بود. مشاهد ههای آغازین گرود نشان داد، این بیماری در خانوادهها تکرار میشود، یعنی اگر یکی از خویشاوندان نسبی این صفت را داشته باشد، احتمال بروز آن در خویشاوندان دیگر نیز هست. اما به طور معمول، والدین کودکان بیمار ، این صفت را ندارند.
مساله: چرا ادرار کودکان دچار شده به آلکاپتونوریا ، پس از برخورد با هوا، تیره میشود؟ آیا این بیماری ارثی است؟ الگوی توارث آن چگونه است؟
گردآوری اطلاعات
در سال 1859، شیمیدانها ثابت کردند در ادرار این بیماران ترکیبی به نام اسید هموجنتیسیک (آلکا پت ون) وجود داردکه پس از واکنش با اکسیژن هوا ، به ماده ی سیاه رنگی تبدیل میشود و ساختمان شیمیایی آن، شبیه ملانین (رنگیز ه ی پوست) است.
چند پژوهشگر مقدار اسید هموجنتسیک ادرار بیماران را اندازهگیری کردند.
|
شماره |
جنسیت |
سن (سال) |
اسید هموجنتیسیک (گرم) |
پژوشگر |
|
1 |
مرد |
5/2 |
2/3 |
اریک مایر |
|
2 |
مرد |
5/3 |
6/2 |
آرچیبلد گرود |
|
3 |
مرد |
8 |
7/2 |
ایوالد استایر |
|
4 |
مرد |
18 |
9/5 |
استانج |
|
5 |
مرد |
44 |
6/4 |
میتلباخ |
|
6 |
مرد |
45 |
7/4 |
اوگدن |
|
7 |
مرد |
60 |
3/5 |
هامارستن |
|
8 |
زن |
60 |
3/2 |
املسن |
|
9 |
مرد |
68 |
8/4 |
ولکاو/ بومن |
توجه: میانگین ترشح اسید هموجنتیسیک به گرم در 24 ساعت و با رژیم غذایی معمولی
پژوهشگران دریافتند :
1. این مولکول در ادرار افراد سالم وجود ندارد
2. همهی بیماران از کودکی این نارسایی را دارند
3. تغییر رژیم غذایی، باعث تغییر مقدار آلکاپتون در ادرار نمیشود
فرضیه سازی
دادههای جدول نشان میدهد، بین سن بیمار ا و میزان ترشح اسید هموجنتیسیک ( و در واقع ، بیماری آلکاپتونوریا ) ارتباطی وجود ندارد و مردها بیش از زنها به این بیماری مادرزادی دچار میشوند. همچنین ، گرود و پژوهشگران دیگر دریافتند، بین این بیماری و رژیم غذایی افراد، ارتباط معناداری وجود ندارد. از این رو، میتوان نتیجه گرفت، این بیماری به اختلالی در واکنشهای شیمیایی بیماران مربوط میشود و مبتلایان این نارسایی را از والدین خود به ارث بردهاند.
فرضیه : اگر این نارسایی ارثی باشد، احتمال بروز آن در بستگان بیمار (برادرها و خواهرها) نیز وجود دارد.
بررسی فرضیه
گرود، برای بررسی این فرضی ه، اطلاعات مربوط به موارد بیماری را که پژوهشگران دیگر گزارش کرده بودند، بررسی کرد.
|
شماره |
تعداد افراد خانواده (برادر و خواهر) |
تعداد مبتلایان به آلکاپتونوریا |
تعداد افراد عادی خانواده |
پژوهشگر |
|
1 |
14 |
4 |
10 |
پاوه |
|
2 |
4 |
3 |
1 |
کایرس |
|
3 |
7 |
3 |
4 |
وینتر نیز |
|
4 |
2 |
1 |
1 |
استایر |
|
5 |
2 |
2 |
0 |
بومن |
|
6 |
1 |
1 |
0 |
مایر |
|
7 |
10 |
1 |
9 |
نوکیولی |
|
8 |
5 |
2 |
3 |
گرود |
|
9 |
3 |
2 |
1 |
گرود |
|
جمع |
48 |
19 |
29 |
|
پرسش1 : دادههای جدول را چگونه تفسیر میکنید؟ آیا این دادهها فرضی هی گرود را ثابت میکنند؟
الکاپتون از کجا میآید
بررسیهای گرود نشان داد، آلکاپتونوریا یک بیماری ارثی است. اما چرا در ادرار مبتلایان مقدار زیادی آلکاپتون وجود دارد؟ در آن زمان، شیمیدانهایی که دربار هی واک نشهای زیست شیمیایی مطالعه میکردند، میدانستند که آنزیمها باعث سرعت این واکنشها میشوند و در نبود آنزیم، این واکنشها انجام نمیشوند. آنان نشان داده بودند که واکنشهایی زیست شیمیایی زنجیره و ا ر انجام میشوند؛ به طوری که ماد هی آغازین ، نخست به چند ماد هی میانی و سپس به فراورده تبدیل میشود. هر کدام از مرحل ههای یک زنجیر هی زیست شیمیایی را یک آنزیم پیش میبرد و اگر یکی از آنزیمها وجود نداشته باشند، ماد هی میانی، که آن آنزیم روی آن کار میکند، انباشته میشود و در صورتی که در آب محلول باشد، با ادرار به بیرون راه مییابد .
فرضیه سازی
ساختمان شیمیایی اسید هموجنتیسیک شباهت زیادی به ساختمان شیمیایی اسید آمین هی تیروزین دارد. (شکل زیر ). از این رو به نظر میرسد، اسید هموجنتیسیک از این اسید آمینه به دست میآید . افراد دچار شده به آلکاپتونوریا ، آنزیمی را که ماد هی میانی آلکاپتون را به ماد هی میانی دیگری تبدیل میکند ، ندارند.
فرضیه: اگر این بیماری پیامد کمبود آنزیمی باشد که روی اسید هموجنتیسیک اثر میگذارد، پس میزان فعالیت این آنزیم باید در بیماران پا ی ین باشد.
بررسی فرضیه
شکل زیر فعالیت آنزیم را در سه گروه افراد عادی، والدین بیماران و افراد بیمار نشان میدهد.
پرسش2 : با توجه به شکل، آیا فرضیه ثابت میشود؟ ادرار والدین بیماران ، پس از قرار گرفتن در معرض هوا به رنگ تیره در نمیآید. با وجود این، فعالیت آنزیم در آنان از افراد عادی کمتر است. این حقیقت را چگونه توجیه میکنید؟ اگر به هر سه گروه مقداری اسید هموجنتیسیک بخورانیم، انتظار دارید ترکیب ادرار آنان چه تغییری پیدا کند؟
50 سال طول کشید تا با نمونهبرداری از کبد مشخص شد، فعالیت آنزیمهای درگیر در تجزی هی اسیدآمینههای فنیل آلانین و تیروزین در افراد سالم و بیمار، به جز آنزیم هموجنتیسیک اسید اکسیداز، یکسان است (شکل زیر ). این آنزیم هموجنتیسیک را به مالیل استواستیک تبدیل میکند و کمبود فعالیت آن، باعث انباشته شدن این اسید در سلولها و سرانجام ترشح آن به ادرار میشود.
پرسش 3 : کودک دو سالهای به بیمارستان آورده شده است. مادرش میگوید، او زیاد استفراغ میکند ( به ویژه پس از شیر خوردن ). وزن کودک و میزان تحرکش زیر اندازهی عادی است. موهایش تیرهاند، اما رگههای سفیدی دارند. نتیج هی بررسی ادرار کودک به صورت زیر است.
|
غلظت برحسب میلی مول | ||
|
ماده |
ادرار بیمار |
ادرار عادی |
|
فنیل آلانین فنیل پیرووات فنیل استات |
0/7 8/4 3/10 |
01/0 0 0 |
الف) با توجه به شکل زیر ، این کودک دچار کمبود چه آنزیمی است؟
ب) برای درمان کودک، چه پیشنهادی دارید.
ج) چرا موهای کودک، رگههای سفید دارند؟
پژوهشهای فراکلاسی
1. همان طور که گفته شد، اسید هموجنتیسیک در برخورد با هوا به مادهای سیاه رنگ تبدیل میشود که ساختمان آن شبیه ملانین (رنگیز هی پوست) است. پژوهش کنید، بین سوخت و ساز ملانین و اسیدهموجنتیسیک چه ارتباطی وجود دارد؟
2. آلبینیسم یکی دیگر از نارسایی مادر زادی در سوخت و ساز است. این نارسایی که به کاهش تولید ملانین و در نتیجه سفید شدن مو میانجامد، به علت کمبود فعالیت یکی از آنزیمهای دخیل در تولید ملانین به وجود میآید.
اقتباس ازوبلاگ گروه زیست شناسی شبه جزیره هند
الف) تریزومی 21 ( سندروم داون)( xx، 45یا xy ،45)
یکی از مشهورترین اختلالات کروموزوم که قبلا" به آن منگولیسم می گفتندسندرم داون استlangdon Down اولین شخصی بود که در سال 1866 علائم کلینیکی این بیماری را توضیح داد.
مبتلایان به این بیماری دارای قد کوتاه ، چین خوردگی پرده گوشه چشم (epicanthus) شانه های کوتاه و پهن ، سوراخ بینی گشاد ، زبان بزرگ، همراه با شیار های مخصوص ، دستهای کوتاه هستند.
عقب افتادگی ذهنی از خصوصیات این افراد است ، ولی می توانند تا حدودی تعلیم داده شوند .این افراد یک کروموزوم 21 اضافه دارند( تریزومی 21) می باشند . خطر ایجاد لوسمی در افراد مبتلا 15 بار بیشتر از افراد معمولی است . حدود یک سوم افراد بیمار دارای ناهنجاریهای قلبی می باشند .
حدود یک ششم نوزادانی که مبتلا به این بیماری متولد می شوند در اولین سال زندگی خود از بین می روند. متوسط عمر این افراد حدود 40 سال است.
ب) تریزومی شماره 18
اولین بار توسط ادواردز توضیح داده شد .فراوانی این بیماری 20000/1است.
مبتلایان به این بیماری اکثرا" در سه ماهگی بعد از تولد از بین می روند ولی بعضی از آنها تا پنج سال هم زنده مانده اند علائم شامل مغز کوچک ، عقب افتادگی ذهنی ، کری، پایین بودن موضع گوشها، گره شدن شستها به صورت خالص می باشند، اکثرا" دارای ناهنحاریهای قلبی می باشند . حدود 80% افراد بیمار دخترند .علت اکثر تریزومی ها سن مادر است.
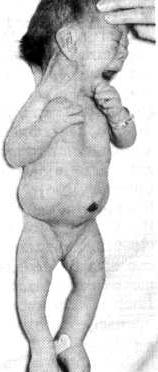
ج) سندرم ترنر (x،44)
سندرم ترنر نوعی مونوزومی است که دارای 44کروموزوم اتوزوم ویک کروموزوم جنسی x می باشند.فنوتیپ این ناهنجاری کروموزومی اولین بار توسط ترنر (H.H.Turner) کشف شد افراد مبتلا دختر های غیز طبیعی هستند که فراوانی آنها 2500/1 در بین متولدین دختر است .بیش از 90% آنها خود به خود سقط می گردند
این گونه زنها فاقد تخمدان هستند و صفات ثانویه جنسی آنها محدود است .از دیگر
عوارض اینگونه افراد قد کوتاه، پایین بودن موضع گوشها ، پایین بودن خط مو در پشت سر ، وجود پرده و چین در گردن ، و سینه های سپر مانند می باشند .افراد مبتلا با مقایسه با برادران وخواهران خود دارای هوش یکسان یاکمی کمتر هستند .
مونوزومی x احتمالا" از تخمک یا اسپرمهای استثنایی بدون کروموزم جنسی یا در اثر حذف کروموزوم جنسی x در اولین تقسیم میتوز بعد از اینکه تخم xxیا xyایجاد شده اند بوجود می آیند . بعضی از افراد مبتلا دارای یک کروموزوم x وقطعه ای از کروموزوم x دوم می باشند .مطالعه ی این گونه افراد نشان داده است که هر دو بازوی کروموزوم x دوم جهت رشد طبیعی تخمدان لازم است .افرادی که فقط بازوی بزرگ x دوم را دارا می باشند قدکوتاه بوده وبقیه عوارض سندرم ترنر را نشان می دهند .افرادی که دارای بازوی کوچک x دوم می باشند دارای قد طبیعی بوده وتمام عوارض را نشان نمی دهند، این موضوع نشان می دهد که فنوتیپ ترنر اکثرا" توسط ژنهای واقع بر روی بازوی کوچک x کنترل می شود.
د) سندرم کلا ین فلتر ( xxy، 44)
اضافه شدن یک کروموزوم xبه ترکیب کروموزومی مرد باعث ایجاد سندرم کلاین فلتر می گردد که اولین بار توسط کلاین فلتر تو ضیح داده شده است. این افراد مذکر بوده و فراوانی آنها 1000/1 در بین متولدین پسر می باشد.
این افراد از نظر فنوتیپی نر ولی در بعضی خصوصیات بخصوص صفات ثانویه جنسی شبیه زنها هستند. برای مثال سینه ها رشد کرده ولی موی بدن رشد نمی کند ، بیضه ها کوچک و غده پروستات کوچک است . علت ایجاد آنها احتمالا" لقاح یک تخمک غیر طبیعی xxبا اسپرم y ، یک تخمک x با اسپرم غیر طبیعی xy است.
حدود سه چهارم افراد بیمار دارای کاریوتیپ ( xxy ، 44) می باشند . ولی در صورتیکه تعداد x ها ویا y ها اضافه شود همین فنوتیپ با اندکی تغیر مشاهده می گردد. برای مثال کاریوتیپهای xxyy , xxxyy,xxxy,xxxxy, ،نیز سندرم کلاین فلتر نامیده می شود.
یکی از عوارض این بیماری عقب افتادگی ذهنی است که بستگی به تعداد x ها دارد،
به این صورت که هر چه تعداد x ها زیاد باشد عقب افتادگی نیز زیادتر می گردد. پسر ها از حد معمول بلند تر بوده ، نسبت قد به وزن آنها طوری است که قد بلند و
باریک به نظر میرسند، قطر سر آنها به طور معنی داری از حد معمول کمتر است،
حالت تعرضی و فعالیت آنها کمتر و حساسیت آنها نسبت به مسائل اجتماعی بیشتر است.
د)هرمافرودیتی در انسان
هرمافرودیت های حقیقی ، افرادی هستند که دارای بافت های تخمدان وبیضه می باشند اعضاء دستگاه تناسلی خارجی مبهم ،ولی اغلب حالت مردانگی کمتری داشته ،خواص ثانویه جنسی از مردانگی تا زنانگی متغیر است . به طور معمول هرمافرودیت حقیقی به علت ناقص بودن اعضا ء تناسلی عقیم اند .هرمافرودیت ناقص ممکن است دارای بافتهای تخمدان یا بیضه باشند .بر مبنای ساختار کروموزومی دو گروه عمده از آنها قابل تشخیص اند که هرمافرودیت کاذب مردانه یا زنانه نامیده می شوند . دستگاه تناسلی خارجی آنها مبهم بوده از نظر ظاهری و صفات ثانویه جنسی با افراد عادی تا حد زیادی دارای فنوتیپ متفاوتی هستند
ه)بیماری سندرم فریاد گربه
در انسان کمبودهای کروموزومی شناخته شده و دارای اثرات مضر می باشد . یکی از این کمبود ها مربوط به بازوی کوچک کروموزوم شماره 5 است که باعث سندر م فریاد گربه ( cri-du- chat )می شود.علت نامگذاری نیز شباهت گریه نوزاد مبتلا به صدای گربه است .از علائم این بیماری می توان میکروسفالی ،افزایش فاصله چشمها،
پایین تر قرار گرفتن گوشها ،مایل شدن چشمها در جهت خلاف داون، کوچکی چانه
( میکروگناسیا)را نام برد
